Part of series:Visualize
GWFM结果可视化
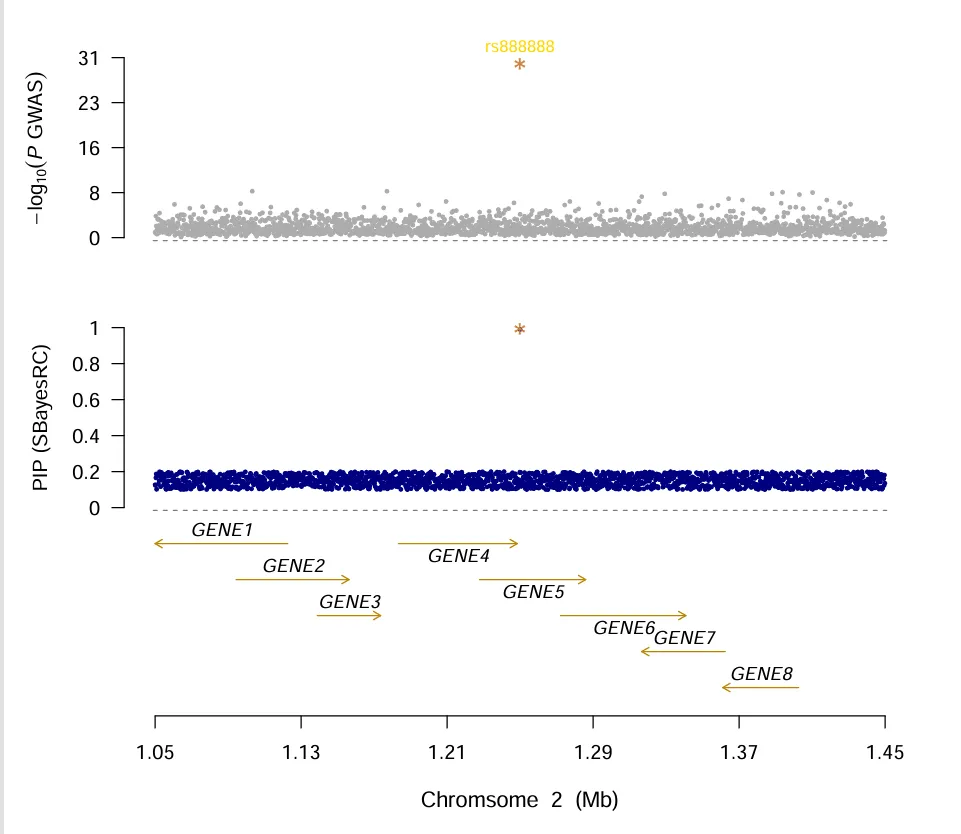
When you want draw locus plot of GWFM, follow scripts will meet your require
plot_GWFM.r
# ===========================================================================================//
# @Author : Loren Shi
# @Time : 2025/06/05 15:42:36
# @File : plot_GWFM.r
# @Mails : crazzy_rabbit@163.com
# @line : https://github.com/Crazzy-Rabbit
#
# R script to draw regional plot for genome wide fine mapping SNPs.
#
# Part of code are adopted from plot_smr script which provided by SMR
#
# Amendment:
# 2025/06/05
# 1. script completed
# 2. first realeased
# 3. fixed teh bug where the drawn gene was not in the middle of the genelayer when nrowgene=1
#
# Usages:
# source("plot_GWFM.r")
# PData <- ReadPvalueFromFiles(gwas="gwas_chrpos.gz", gwfm="gwaf.snpRes", glist="glist_hg19_refseq.txt", windowsize=200000, highlight="rs641221")
# pdf('gwfm_plot.pdf',width = 8,height = 8)
# MultiPvalueLocusPlot(data=PData)
# dev.off()
# ===========================================================================================//
is.installed <- function(mypkg){
is.element(mypkg, installed.packages()[,1])
}
# check if package "TeachingDemos" is installed
if (!is.installed("TeachingDemos")){
install.packages("TeachingDemos");
}
library(TeachingDemos)
library(data.table)
# parmeters for plot
genemove=0.01; txt=1.1; cex=1.3; lab=1.1; axis=1; top_cex=1.2;
GeneRowNum = function(GENELIST) {
BP_THRESH = 0.03; MAX_ROW = 5
# get the start and end position
GENELIST = GENELIST[!duplicated(GENELIST$GENE),]
START1 = as.numeric(GENELIST$GENESTART);
END1 = as.numeric(GENELIST$GENEEND)
STRLENGTH = nchar(as.character(GENELIST$GENE))
MIDPOINT = (START1 + END1)/2
START2 = MIDPOINT-STRLENGTH/250;
END2 = MIDPOINT+STRLENGTH/250
START = cbind(START1, START2);
END = cbind(END1, END2);
START = apply(START, 1, min);
END = apply(END, 1, max)
GENELIST = data.frame(GENELIST, START, END)
GENELIST = GENELIST[order(as.numeric(GENELIST$END)),]
START = as.numeric(GENELIST$START); END = as.numeric(GENELIST$END)
# get the row index for each gene
NBUF = dim(GENELIST)[1]
ROWINDX = rep(1, NBUF)
ROWEND = as.numeric(rep(0, MAX_ROW))
MOVEFLAG = as.numeric(rep(0, NBUF))
if(NBUF>1) {
for( k in 2 : NBUF ) {
ITERFLAG=FALSE
if(START[k] < END[k-1]) {
INDXBUF=ROWINDX[k-1]+1
} else INDXBUF = 1
if(INDXBUF>MAX_ROW) INDXBUF=1;
REPTIME=0
repeat{
if( ROWEND[INDXBUF] > START[k] ) {
ITERFLAG=FALSE
INDXBUF=INDXBUF+1
if(INDXBUF>MAX_ROW) INDXBUF = 1
} else {
ITERFLAG=TRUE
}
if(ITERFLAG) break;
REPTIME = REPTIME+1
if(REPTIME==MAX_ROW) break;
}
ROWINDX[k]=INDXBUF;
if( (abs(ROWEND[ROWINDX[k]]-START[k]) < BP_THRESH)
| ((ROWEND[ROWINDX[k]]-START[k])>0) ) {
MOVEFLAG[k] = 1
SNBUF = tail(which(ROWINDX[c(1:k)]==ROWINDX[k]), n=2)[1]
MOVEFLAG[SNBUF] = MOVEFLAG[SNBUF] - 1
}
if(ROWEND[ROWINDX[k]]<END[k]) {
ROWEND[ROWINDX[k]] = END[k] }
}
}
GENEROW = data.frame(as.character(GENELIST$GENE),
as.character(GENELIST$ORIENTATION),
as.numeric(GENELIST$GENESTART),
as.numeric(GENELIST$GENEEND),
ROWINDX, MOVEFLAG)
colnames(GENEROW) = c("GENE", "ORIENTATION", "START", "END", "ROW", "MOVEFLAG")
return(GENEROW)
}
ReadPvalueFromFiles <- function(gwas, gwfm, glist, windowsize=500000, highlight) {
gwas1 = fread(gwas)[, .(CHR, POS, SNP, p)]
colnames(gwas1) = c("CHR", "BP", "SNP", "p")
gwas1$CHR = as.character(gwas1$CHR)
gwas2 = fread(gwfm)[, .(Chrom, Position, Name, PIP)]
colnames(gwas2) = c("CHR", "BP", "SNP", "p")
gwas2$CHR = as.character(gwas2$CHR)
snp_gwas1 = gwas1[gwas1$SNP == highlight, ];
chrom = unique(snp_gwas1$CHR)
if (nrow(snp_gwas1) == 0) {stop("highlight SNP not found in file1, please check it!")}
BP1 = snp_gwas1$BP
start1 = BP1 - windowsize;
end1 = BP1 + windowsize;
file1 = gwas1[gwas1$CHR == chrom & gwas1$BP > start1 & gwas1$BP < end1, ]
snp_gwas2 = gwas2[gwas2$SNP == highlight, ];
if (nrow(snp_gwas2) == 0) {stop("highlight SNP not found in file2, please check it!")}
BP2 = snp_gwas2$BP
start2 = BP2 - windowsize;
end2 = BP2 + windowsize;
file2 = gwas2[gwas2$CHR == chrom & gwas2$BP > start2 & gwas2$BP < end2, ]
start = max(c(start1, start2), na.rm=T) / 1e6;
end = max(c(end1, end2), na.rm=T) / 1e6;
glist = fread(glist)
glist[,2] = glist[,2]/1e6;
glist[,3] = glist[,3]/1e6;
colnames(glist) = c("CHR", "GENESTART", "GENEEND", "GENE", "ORIENTATION")
glist = glist[glist$CHR == chrom & glist$GENESTART >= start & glist$GENEEND <= end, ]
return_lsit = list(file1=file1, file2=file2, SNP=highlight, glist=glist, CHR=chrom)
}
MultiPvalueLocusPlot <- function(data) {
gwas1 = data$file1;
gwas2 = data$file2;
pXY1 = -log10(gwas1$p);
yMAX = ceiling(max(pXY1, na.rm=T)) + 1;
pXY2 = gwas2$p;
yMAX2 = ceiling(max(pXY2, 1, na.rm=T));
glist = data$glist;
generow = GeneRowNum(glist);
num_row = max(as.numeric(generow$ROW));
offset_map = ceiling(yMAX);
offset_map = max(offset_map, num_row*2.5);
offset_pip = yMAX / 2.5;
dev_axis = 0.1*yMAX;
if (dev_axis < 1.5) dev_axie=1.5;
yaxis.min = -offset_map - offset_pip - dev_axis - yMAX;
yaxis.max = yMAX + ceiling(offset_pip) + 1;
gwasBP1 = as.numeric(gwas1$BP) / 1e6;
gwasBP2 = as.numeric(gwas2$BP) / 1e6;
xmin = min(c(gwasBP1, gwasBP2), na.rm=T) - 0.001;
xmax = max(c(gwasBP1, gwasBP2), na.rm=T) + 0.001;
xlab = paste("Chromsome ", data$CHR, " (Mb)")
#------------------- plot gwas layer ----//
ylab1 = expression(-log[10] (italic(P) * " GWAS"))
par(mar=c(5,5,3,2), xpd=TRUE);
plot(gwasBP1, pXY1, pch=20, xaxt="n", yaxt="n", bty="n", ylim=c(yaxis.min, yaxis.max),
xlim=c(xmin, xmax), xlab=xlab, ylab="", cex.lab=lab, cex.axis=axis,cex=0.6, col="gray68");
devbuf1 = yMAX/4;
xticks = round(seq(xmin, xmax, length.out=6), 2);
axis(1, at=xticks, lwd=1);
axis(2, at=seq(0, yMAX, by=devbuf1), labels=round(seq(0, yMAX, devbuf1), 0), las=1, lwd=1);
mtext(ylab1, side=2, line=3, at=(yMAX*1/2));
segments(x0=xmin, y0=-0.5, x1=xmax, y1=-0.5, col="grey50", lwd=1, lty=2);
snp1 = gwas1[SNP == data$SNP, ]
snpBP1 = snp1$BP / 1e6;
snpP1 = -log10(snp1$p);
points(snpBP1, snpP1, pch="*", col="peru", cex=2);
text(snpBP1, snpP1, labels=snp1$SNP, col="gold", pos=3, cex=0.8, offset=0.5);
#------------------- plot PIP layer ----//
ylab2 = "PIP (SBayesRC)"
axis.start = 0;
axis.start = axis.start - yMAX - offset_pip - dev_axis;
pXY2buf = pXY2 / yMAX2*yMAX + axis.start;
par(new=TRUE);
plot(gwasBP2, pXY2buf, pch=20, xaxt="n", yaxt="n", bty="n", ylim=c(yaxis.min, yaxis.max),
xlim=c(xmin, xmax), ylab="", xlab="", cex.lab=lab, cex.axis=axis, cex=0.6, col="navy");
devbuf2 = yMAX2/5;
axis(2, at=seq(axis.start, (axis.start+yMAX), yMAX/5), labels=seq(0, yMAX2, devbuf2), las=1, lwd=1);
mtext(ylab2, side=2, line=3, at=((axis.start + axis.start + yMAX)/2));
segments(x0=xmin, y0=axis.start-0.5, x1=xmax, y1=axis.start-0.5, col="grey50", lwd=1, lty=2);
snp2 = gwas2[SNP == data$SNP, ]
snpBP2 = snp2$BP / 1e6;
snpP2 = snp2$p / yMAX2*yMAX + axis.start;
points(snpBP2, snpP2, pch="*", col="peru", cex=2);
#------------------- plot gene layer ----//
num_gene = dim(generow)[1]
dist = offset_map / num_row;
for (k in 1:num_row) {
generowbuf = generow[which(as.numeric(generow[, 5]) == k), ]
xstart = as.numeric(generowbuf[, 3])
xend = as.numeric(generowbuf[, 4])
snbuf = which(xend - xstart < 1e-3)
if (length(snbuf) > 0) {
xstart[snbuf] = xstart[snbuf] - 0.0025
xend[snbuf] = xend[snbuf] + 0.0025
}
xcenter = (xstart+xend)/2
xcenter = spread.labs(xcenter, mindiff=0.01, maxiter=1000, min=xmin, max=xmax)
num_genebuf = dim(generowbuf)[1]
if (num_row == 1) {
for (l in 1:num_genebuf) {
ofs=0.3;
if (l%%2 == 0) ofs=-0.8;
ypos = offset_map/2 + yaxis.min - 1;
code = 1;
if (generowbuf[l,2] == "+") code=2;
arrows(x0=xstart[l], y0=ypos, x1=xend[l], y1=ypos, code=code, length=0.07, ylim=c(yaxis.min, yaxis.max),
col=colors()[75], lwd=1)
movebuf = as.numeric(generowbuf[l, 6]) * genemove
text(x=xcenter[l]+movebuf, y=ypos, label=substitute(italic(genename), list(genename=as.character(generowbuf[l,1]))), pos=3, offset=ofs, col="black", cex=0.9)
}
} else if (num_row > 1) {
for (l in 1:num_genebuf) {
ofs=0.3
if(l%%2==0) ofs=-0.8
m = num_row - k
ypos = m*dist + yaxis.min
code = 1;
if (generowbuf[l,2] == "+") code = 2;
arrows(x0=xstart[l], y0=ypos, x1=xend[l], y1=ypos, code=code, length=0.07, ylim=c(yaxis.min, yaxis.max),
col=colors()[75], lwd=1)
movebuf = as.numeric(generowbuf[l,6]) * genemove
text(x=xcenter[l]+movebuf, y=ypos, label=substitute(italic(genename), list(genename=as.character(generowbuf[l,1]))), pos=3, offset=ofs, col="black", cex=0.9)
}
}
}
}example data
set.seed(123)
n_snps <- 10000
n_genes <- 10
## SNP pos
positions <- as.integer(seq(500000, 2500000, length.out = n_snps))
snp_ids <- paste0("rs", 1000000 + 0:(n_snps - 1))
## highlight SNP(GWAS)
highlight_snp <- data.frame(
CHR = "2",
POS = 1250000,
SNP = "rs888888",
p = 1e-30
)
## GWAS summary
pvals <- pmax(rbeta(n_snps, 0.4, 5), 1e-8)
gwas_df <- data.frame(
CHR = rep("2", n_snps),
POS = positions,
SNP = snp_ids,
p = pvals
)
gwas_df <- rbind(gwas_df, highlight_snp)
write.table(gwas_df, file = "sim_gwas_chr2.gz", sep = "\t", row.names = FALSE, quote = FALSE)
## highlight SNP(PIP)
highlight_pip <- data.frame(
Chrom = "2",
Position = 1250000,
Name = "rs888888",
PIP = 0.99
)
# PIP data, finemapping result
pip_values <- round(runif(n_snps, 0.1, 0.2), 4)
gwfm_df <- data.frame(
Chrom = rep("2", n_snps),
Position = positions,
Name = snp_ids,
PIP = pip_values
)
gwfm_df <- rbind(gwfm_df, highlight_pip)
write.table(gwfm_df, file = "sim_gwfm.snpRes", sep = "\t", row.names = FALSE, quote = FALSE)
# gene list
gene_starts <- as.integer(seq(1050000, 1450000, length.out = n_genes))
gene_lengths <- sample(30000:80000, n_genes, replace = TRUE)
gene_ends <- gene_starts + gene_lengths
gene_df <- data.frame(
CHR = rep("2", n_genes),
GENESTART = gene_starts,
GENEEND = gene_ends,
GENE = paste0("GENE", 1:n_genes),
ORIENTATION = sample(c("+", "-"), n_genes, replace = TRUE)
)
write.table(gene_df, file = "sim_glist_chr2.txt", sep = "\t", row.names = FALSE, quote = FALSE)run script
source("plot_GWFM.r")
PData <- ReadPvalueFromFiles(gwas="sim_gwas_chr2.gz", gwfm="sim_gwfm.snpRes", glist="sim_glist_chr2.txt", windowsize=200000, highlight="rs888888")
pdf('gwfm_plot.pdf',width = 8,height = 8)
MultiPvalueLocusPlot(data=PData)
dev.off()